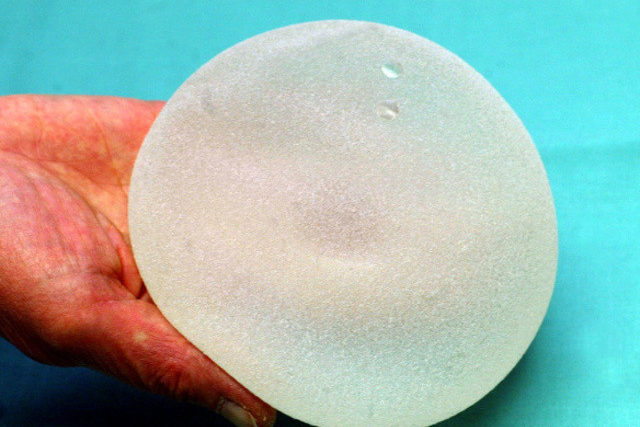
Die EU-Kommission will die Kontrollen für Medizinprodukte verschärfen. Die strikteren Vorschriften sind eine Folge des Skandals um fehlerhafte Brustimplantate des französischen Herstellers PIP.
Der für Gesundheit zuständige EU-Kommissar John Dalli sagte bei der Vorstellung der Vorschläge am Mittwoch in Brüssel, künftig sollten die zuständigen nationalen Behörden verstärkt unangekündigte Fabrikbesuche und Stichproben bei Herstellern durchführen können. Bisher seien solche Inspektionen den Produzenten zwei Monate im Voraus gemeldet worden, eine solche Ankündigung soll nun entfallen.
„Ich glaube, dass diese Regeln nicht nur die Sicherheit der Konsumenten gewährleisten werden, sondern auch das Vertrauen zurückbringen“, sagte Dalli. Der EU-Kommissar betonte, im Fall von PIP sei nicht die Marktzulassung des Produktes der entscheidende Punkt gewesen. Vielmehr habe es sich um Betrug der Firma gehandelt.
Die neuen Vorschriften in der EU haben indirekt auch Auswirkungen auf die Schweiz, über das bilaterale Abkommen zur „gegenseitigen Anerkennung von Konformitätsbewertungen“. Bezüglich des Imports von Medizinprodukten und In-vitro-Diagnostika aus einem Drittstaat in die EU hält die Kommission fest, dass die „gleichen Vorschriften gelten“ wie für die in der EU hergestellten Produkte.
Neue Datenbank
Ein europäisches Experten-Panel mit Vertretern aus jedem EU-Staat soll sicherstellen, dass die Prüf-Standards EU-weit eingehalten werden, sagte Dalli. Zur besseren Koordinierung der EU-Staaten schlug die EU-Kommission auch die Schaffung einer zentralen europäischen Datenbank für Medizinprodukte (Eudamed) vor.
Parallel zum neuen Rechtsrahmen für Medizinprodukte präsentierte die EU-Kommission auch neue Vorschriften für In-vitro-Diagnostika wie Schwangerschafts- oder HIV-Tests. Der Geltungsbereich der bisherigen Richtlinie soll etwa auf Implantate für kosmetische Zwecke ausgeweitet werden.
Um Rückrufaktionen gegebenenfalls leichter durchführen zu können, will die EU-Kommission die Rückverfolgbarkeit der Produkte besser sicherstellen. In-vitro-Diagnostika sollen künftig in vier Risikoklassen eingeteilt werden, wie dies bei anderen Medizinprodukten schon der Fall ist.
Über die Vorschläge der EU-Kommission entscheiden das EU-Parlament und der Ministerrat (Vertreter der EU-Mitgliedstaaten). Er hoffe, dass die Änderungen rasch beschlossen werden können, sagte Dalli.